Studying Sleep the High-Throughput Way
/
“Sleep remains one of the least understood phenomena in biology,” reads the first sentence of a recent review in Cell (1). Though humans spend a third of their life sleeping, neurobiologists don’t really understand how or why we sleep. The scientific method proceeds from a hypothesis, and formulating a hypothesis requires some initial information, which is not really there for sleep. What can scientists do to gather this initial information? A favorite approach of molecular biologists over the past forty years has been the high-throughput screen, a fancy term for trying a bunch of things and seeing if they affect the process of interest. Until recently, it was impractical in neurobiology because it was too hard to collect and analyze the required large amounts of data. However, advances in computing power and the availability of a certain device called the Zebrabox, which I’ll explain later, made it possible for Jason Rihel and colleagues to apply the high-throughput screen to the neurobiology of sleep (2). To paraphrase from that paper’s abstract, the authors set out to find new drugs that affect sleep and to discover if any known proteins have a previously unknown effect on sleep. I am not a sleep expert or even a neurobiologist, but my background in systems biology has taught me a thing or two about high-throughput screens. Below I will explain what makes a good high-throughput screen, what Rihel and colleagues have accomplished, and what they could have done better. A good high-throughput screen generates hypotheses. It fills the initial void of knowledge with information that can be used to perform more targeted experiments. To perform a screen, biologists set up an experiment that reduces what they are studying to some measurement, change one thing in their experiment and make the measurement, change another thing and make the measurement, and repeat this hundreds or even thousands of times. To keep themselves from going crazy, they try to set up a simple measurement, so that repeating it ad nauseam would not be too tedious, time-consuming, or labor- and resource-intensive. However, the measurement shouldn’t be so simple that it doesn’t relate back to what is being studied. A classic example of a smashingly successful screen is the work of Lee Hartwell and colleagues on cell division cycle mutants in budding yeast in the 1970s (3). They were studying how cells divide. A yeast cell assumes a sequence of distinctive shapes as it divides, so they reduced cell division to whether a cell has a normal or abnormal shape. The things that they were changing were genes. By mutagenizing yeast, examining the shape of the resulting cell, and then mapping the mutant locus, they discovered many genes that affected cell shape. One of their hits, named cdc2, was revealed by subsequent targeted experiments to be the master regulator of how cells of all eukaryotic model organisms divide and is intensively studied to this day. Without the screen for yeast mutants of cell shape, no one may have ever connected this particular gene with the cell division cycle.
What did Rihel and colleagues do in their screen? First, they defined sleep simply enough to make it amenable to screening. Until a decade ago, the scientific definition of sleep was an altered state of electrical activity in the brain, as measured by sticking electrodes onto the scalp (1, 4, 5). Sticking electrodes to scalps is fine for making a handful of measurements, but doing it enough times for a screen is impractical, especially since the animals involved, i.e. primates, monkeys, rats, mice, or birds, are relatively large and expensive to maintain. Rihel and colleagues used the inexpensive and easy-to-maintain zebrafish. They defined sleep as lack of movement, following a push begun in the 1980s to define sleep as a behavior (1). Since, they were looking for new drugs, the things they changed were chemicals that they added to the aquarium water. Detecting lack of movement may seem like a simple measurement to make, but it’s not. Back in Lee Hartwell’s days, some poor grad student would have actually been watching the zebrafish or movies of zebrafish for inactivity. Luckily, technology has progressed, and Rihel and colleagues were able to buy a big blue box called the Zebrabox sold by a company named Viewpoint. The Zebrabox is equipped with a 96-well-plate holder, a video camera, and custom video processing software called Videotrack. Rihel and colleagues placed their more than 50,000 zebrafish larvae into 96-well plates, added one of over 5000 chemicals into each well, popped the plates inside the Zebrabox, recorded movies, and analyzed these movies for lack of motion. The chemicals that made zebrafish move more or less were considered hits. The targets of the chemicals, gleaned from annotations in databases and manual literature searches, were by extension implicated in sleep regulation.
The Zebrabox
[youtube]http://www.youtube.com/watch?v=ot48aM8Isvk[/youtube]
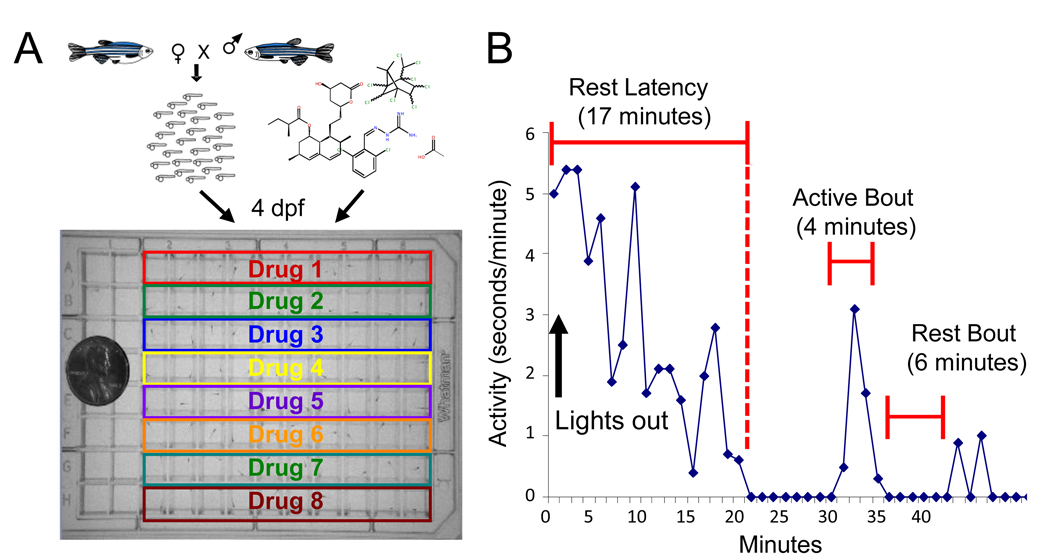
So, how does the screen performed by Rihel and colleagues do in terms of generating hypotheses? Some chemicals they tested, including nicotine and mefloquine (see my previous post for more on this drug) make zebrafish move differently. However, their results have little credibility because they do not justify why lack of motion in zebrafish is definitely sleep, and not tiredness or death. Also, it is debatable how good the drug target annotations are. Some of the more surprising new sleep regulators, like inflammatory cytokines, may be genuine. Or they may be just the only annotated targets of drugs, while the effect of a drug on sleep may be due to some side effect unrelated to the annotated target. I hope that the hits are all genuine and that this work leads to new insights in sleep. But tellingly, a review of recent sleep literature (1) focused on how Rihel and colleagues confirmed what has already been known, rather than on how they may have discovered something new.
Part of the problem with credibility has to do with their black-box, or rather blue-box, approach. They put zebrafish into the Zebrabox and out came all their data for the Science paper. Without knowing how the video processing software Videotrack works, and without a positive control of a drug that is known to make zebrafish move a lot and a negative control of a drug that is known to sedate them, I can only trust that Videotrack gave Rihel and colleagues the result that they claim. Automation can make previously impossible experiments possible, but if the results are ambiguous and untrustworthy, it’s of little value.
In summary, Rihel and colleagues applied high-throughput screening, responsible for groundbreaking discoveries in other areas of biology, to sleep. Notably, they were able to use a complicated measurement of zebrafish behavior because they used an automated measurement and analysis device. But the automated device also robbed their results of credibility. Thus, their paper makes me wish for someone to do a similar screen but in a more transparent way and with a more precise experimental definition of sleep. Then we could make some hypotheses and kick-start the scientific method in the study of sleep.
Sources
- Sehgal A and E Mignot. (2011). “Genetics of Sleep and Sleep Disorders.” Cell. 146:194-207. Paywall.
- Rihel J et al. (2010). “Zebrafish Behavioral Profiling Links Drugs to Biological Targets and Rest/Wake Regulation.” Science. 327:348-351. Paywall.
- Hartwell LH et al. (1973). “Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants.” Genetics. 74: 267-286. Open access.
- Zimmerman JE et al. (2008). “Conservation of sleep: insights from non-mammalian model systems.” Trends in Neurosciences. 31:371-376. Paywall.
- http://en.wikipedia.org/wiki/Electroencephalography