A series of papers from Kang Shen's lab, which I have recently joined, sheds light on a key and fundamental step in the process of transporting pre-synapse forming proteins down the axon and forming functional synapses at exactly the right locations. Here, I’ll be reviewing the first in this series, by Klassen and colleagues, from 2010.
Neurons communicate with each other by sending electrical signals down axons, and across synapses to target other neurons. These synapses along and at the ends of axons can be extremely far away from the cell body, where most of the proteins are created. So both transporting synaptic proteins down the axon and forming synapses at the correct locations are two formidable challenges for the developing neuron. A 2010 paper from Kang Shen’s lab shows that these two processes appear to be intricately linked. The paper provides key evidence that instead of pre-synaptic proteins being transported in separate pieces, and assembled from scratch onsite as a functioning synapse, all the major protein components of the pre-synapse are transported together, ready for quick and easy assembly upon arrival. The paper shows that Arl-8 is the clasp that keeps a lid on the loaded-spring like capacity of these pre-synapse cargos that are in transit, preventing them from jumping off the transport train and assembling functional synapses.
Each neuron in the brain connects to only a small subset of the other neurons in the brain, and the selection of the appropriate target neurons is crucial to forming a well functioning brain. After an axon has reached its target destination, it will connect with other neurons in the target area by forming two types of synapses. The axon can form terminal synapses, which are the synapses at the very ends of axons, and which are depicted below at the very tips of the axon branches, or the axon can form en passant synapses, which are the bud-like bright spots along the axon, both of which are depicted below in this image of an axon that is targeting the monkey visual cortex.
In both cases, there are two fundamental challenges that the neuron needs to solve. 1) Neurons somehow need to transport all these synapse-forming proteins from the cell body down the axon to the pre-synaptic specializations in the target area, either to terminal synapses or en passant synapses. 2) Once in the target area, the synapse-forming proteins somehow need to form the right number of en passant and terminal synapses, and in the right locations too! What a fantastically complicated cell biology problem! Yet, somehow, amazingly, evolution has come up with mechanisms to enable these synapses to form in their correct locations.
Now, how to go about deciphering these mechanisms of axon transport and synapse formation? In the mammalian brain, most neurons are very complicated; they send their axons very long distances in the brain through an absolute forest of dense axon bundles, only to arrive at a destination, composed of cell bodies and their dendritic trees that are just as dense and complex. However, many of the same cell biological mechanisms at work in the mammalian brain are also present to a similar extent in the brains of simpler creatures as well, such as the tiny worm known as C. Elegans, which has only 302 total neurons. One of the C. Elegans neurons, the DA9 motoneuron makes exactly 25 en passant synapses along its single, unbranched axon as it courses along the dorsal nerve cord (shown in Panel A, below), and is an excellent model to study these questions of synapse assembly.

Now, in order to begin to understand the mechanisms of pre-synapse axonal transport and formation, one needs to be able to examine the roles of individual proteins in these processes. By disrupting one protein building block in this synapse-formation process, one at a time, we can see what role each of these protein actors plays in the exquisite biological “production” of assembling a pre-synapse. Klassen and Shen therefore created a bunch of mutant worms by chemically inducing mutations in worms. They then took these mutant worms and examined the pattern of distribution of a specific pre-synaptic protein called Rab-3 in the DA9 axon, and if there were any irregularities in the Rab-3 distribution, they would examine the worm’s genome to find the mutant gene, whose defect was responsible for the irregular distribution.
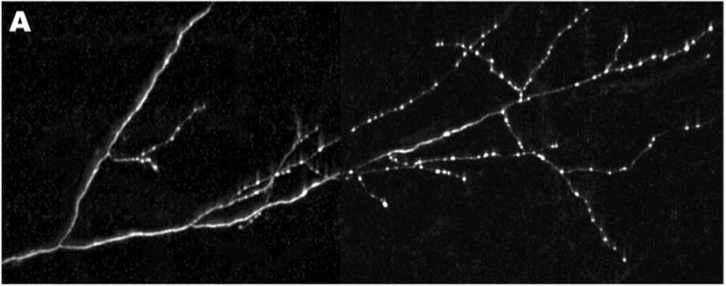
Rab-3 is a protein, which closely associates with small bubbles of membrane at the pre-synaptic specialization containing packets of neurotransmitter, called synaptic vesicles, and helps release these vesicles so they can travel across the synapse. Rab-3 is present in small amounts all along the axon, but large, bright clusters of Rab-3, which are visible in white, occur at sites where synaptic vesicles accumulate. Such vesicle accumulation indicates the presence of a presumptive pre-synaptic site.
By keeping track of where Rab3 clusters would form, Klassen and Shen could examine different mutants to see where the pattern of synaptic vesicle clusters appeared differently from the evenly spaced 25 clusters that normally form along the middle of the axon. Using this strategy, they were able to isolate a mutant worm, where the Rab-3 marked vesicle clusters formed too close to the cell body (Panel C, bottom), and did not get far enough down the axon to form the 25 synapses along the axon like they did in normal worms (Panel B, middle). This mutant had a defective gene encoding a small protein called Arl-8.
In the axons of Arl-8 mutants, the Rab-3 clusters were found to be located very close to the cell-body of the neuron, and much fewer of these clusters were found toward the middle and the end of the axon. It was as if all the synaptic vesicle proteins, marked by the presence of Rab-3, jumped off the transport-train way too early along the axon in the Arl-8 mutant worms. Thus, Arl-8 seems necessary to prevent premature aggregation of Rab-3, and to ensure proper transport of synaptic vesicle associated proteins.
Now, the previously discussed evidence implicated Arl-8 in preventing the aggregation of one of the two major classes of pre-synaptic proteins, the vesicle-associated proteins. As explained earlier, Rab-3 is a member of the class of pre-synaptic proteins called synaptic transport vesicle proteins, which play a role in the primary function of the pre-synapse. Yet, there is an entirely different class of proteins, called active zone proteins, which are a bunch of sticky organizing proteins that collectively form the structural backbone of the pre-synapse. One can think of active zone proteins as being like the engineers and construction workers, who assemble and provide structural support for a missile battery, while the synaptic vesicle release proteins can be thought of as an entirely different group: the soldiers who operate the missile machinery, helping set off the fuse.
Previously, it was thought that each of the two different kinds of proteins were transported down the axon separately from one another. So, all the proteins in the active zone group of proteins would be transported together in the same vesicles, and all of the vesicle associated proteins, like Rab-3, would be transported together, in a class of vesicles that were completely separate from those that transported vesicle associate proteins. In other words, people thought that the engineers travelled to the site of missile assembly in one railroad car, and that the soldiers travelled to the missile site in a totally separate railroad car. However, a second finding concerning Arl-8 challenged this theory that both sets of proteins are transported separately, and then jointly assembled at the synapse.
The second finding is that when one of these sticky, active zone proteins is mutated in addition to Arl-8, the Rab-3 aggregates in the DA9 axons do not have as severe a defect in distribution along the axon. In fact, these double-mutants had a pattern of Rab-3 puncta that looked closer to that of the normal worms, with little bright dots spread out along the entire axon. Klassen and Shen inferred that these sticky active zone proteins were causing Rab-3, and other synaptic vesicle associated proteins to aggregate and cluster together. The fact that mutations in these sticky, clustering, active zone proteins result in less Rab-3 synaptic puncta getting ‘caught’ early in the axon, suggests that both classes of proteins are actually transported together.
This finding provided evidence that instead of transporting the active-zone pre-synapse backbone proteins in certain types of vesicles, and separately transporting vesicle associated proteins in other vesicles, then assembling an entire synapse once all the packaged material arrived onsite, that in fact both types of vesicles were transported together. Or, to return to our analogy, the engineers, military men, and all the components of a mobile missile assembly are transported to a target site together. So, in essence, all the components of the pre-synapse are transported together, ready for quick assembly and deployment when they reach the correct spot.
The reason that this idea of co-transport is cool is that it totally changes the way we might think about how pre-synapses are set up. Instead of building a pre-synapse from scratch each time the neuron wants to form a connection to another neuron, and having to set up all the right active zone and neurotransmitter vesicle release proteins onsite, there might be partially pre-assembled synaptic machinery that’s transported down the axon. And then, when these transported vesicles get to the right place, they are immediately ready to spring into action and form a fully functioning synapse. Arl-8 then is the clasp that prevents these pre-synaptic proteins that are all being transported together from spontaneously aggregating and springing into action to form a synapse too early down the length of the axon.
An immediately exciting future question that is suggested by this research is how Arl-8 might interact with different proteins that set up synapses in particular locations. Perhaps there are other proteins that inhibit the action of Arl-8, in effect releasing this clasp on synapse formation? Perhaps also, there are other proteins, which counterbalance Arl-8, and actually promote the clustering of pre-synaptic proteins and the formation of synapses? Is Arl-8 part of some master switch, which is modulated to set up pre-synapses at specific locations in the brain? If so, then discovering other proteins, which interact with Arl-8, could give us clues into questions like how an axon from the part of the brain that responds to vision knows to form lots of synapses onto face-processing neurons, and not other irrelevant neurons located close by.





 In this edition of Ask a Neuroscientist, we’ll answer two questions that address a similar principle: Can you train to have a better brain?
In this edition of Ask a Neuroscientist, we’ll answer two questions that address a similar principle: Can you train to have a better brain?