
Inflammatory comments on ageing
/
None of us is getting any younger. In fact, most of us can expect to live longer than ever. The world’s oldest man died just last week aged 116 and, as global life expectancy continues to increase, we’re going to have to tackle age-related frailty and functional fragility to avoid unbearable healthcare costs and provide quality of life for a growing population of centenarians. With rates of dementia as high as 60% in the over 85s1, neurodegeneration is a big issue; but treating age-related diseases in isolation may not be the most effective solution. Scientists now think of ageing as an intricate process during which tissues and cells change the way they communicate and interact, resulting in a gradual loss of function. Not exactly a simple cause-effect pairing that can be hit with a youth pill.
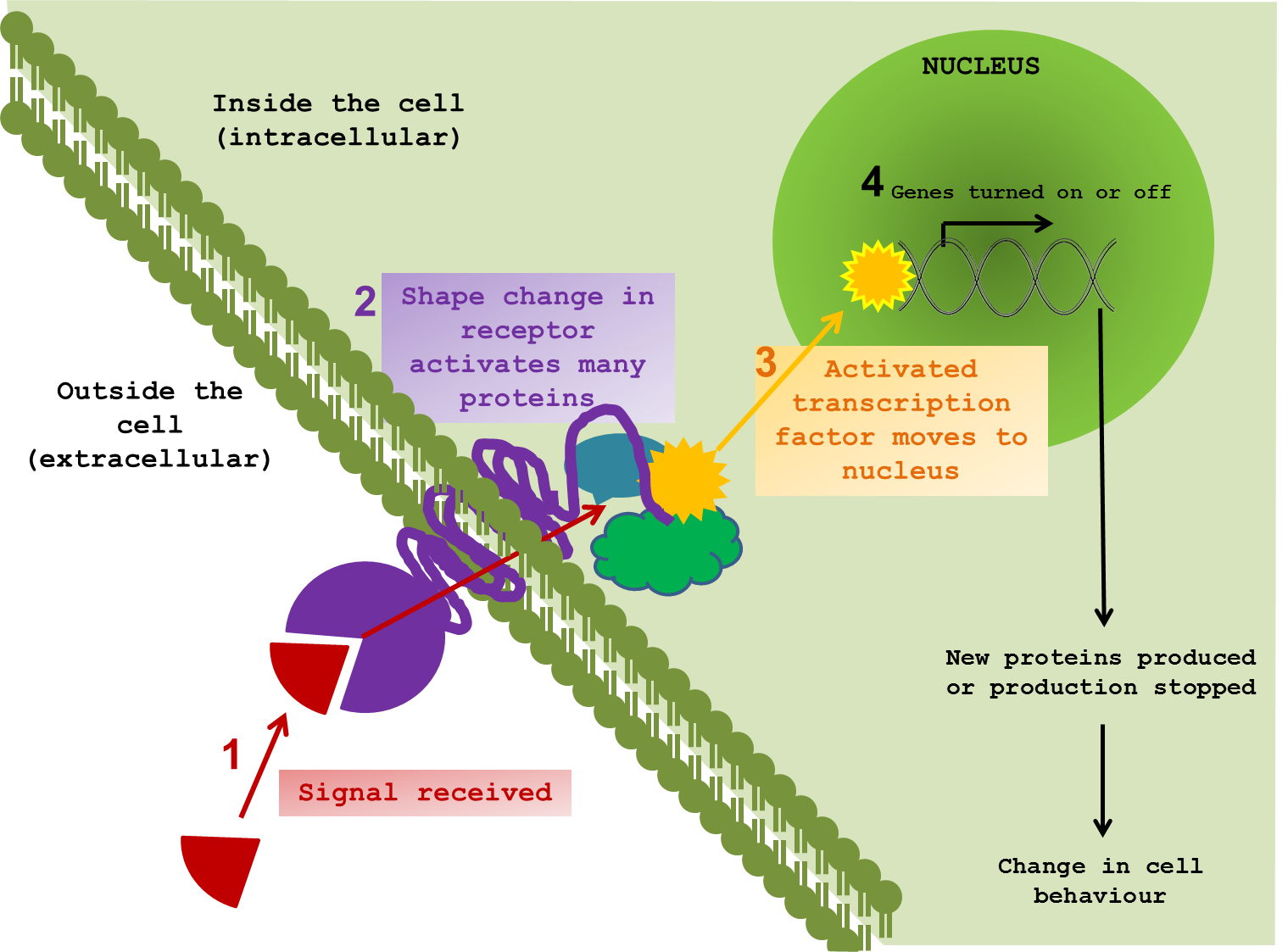
So how can we even start to pick apart the elaborate tapestry of ageing? What exactly happens when a cell changes the way it communicates? To perform effectively, neurons need to pick up signals from the outside world. Indeed all cells, tissues and organs function by responding to external signals, thus allowing an organism to respond and adapt to changes in environment. It is difficult to show in a static diagram the dynamic nature of these interactions. But I tried anyway. ↓

The majority of signals are chemical compounds, floating around outside a cell, waiting to contact their receptor protein. Once in range, the signal and the receptor are drawn closer and dock together. In response to the signal, the receptor twists and changes; not only on the surface, but also through the cell membrane and into the cytoplasm – the inner world of the cell. The shifting and gyrating receptor can hit other proteins, slapping on new chemical groups, removing others and sending these messengers on through the internal cellular space on a collision course with target proteins of their own. This molecular line of dominoes almost always ends at the cell’s control centre – the nucleus. It is here that messages are converted into changes in gene expression, protein production and, in turn, cellular behaviour. The proteins that directly interact with DNA to bring about such changes are called transcription factors.
Different signal molecules and receptors trigger distinct domino effects in the cell, which scientists refer to as signalling pathways. Though different cells are tuned to pick up different signals, many of the intracellular proteins used to transmit the message have been the same for millions of years. Throughout evolution, cells have taken the Heath Robinson* approach to signalling. Rather than inventing new proteins every time a new function had to be performed, cells used the tools already at their disposal. As a result, the same proteins can be found in different types of cells, across species and even across kingdoms. You share molecular signalling pathways with almost everything – from bears, to bananas to bacteria.
*Americans see Rube Goldberg
One versatile and highly abundant transcription factor, and the focus of a new study in Nature2, is NFkB (nuclear factor kappa B). This molecule is a hallmark of inflammation and once activated, perpetuates inflammatory responses. However, many different cells use this molecule to transmit all sorts of messages and, as a consequence, its role in a particular signalling pathway is often difficult to pin down. In their new study, Zhang and colleagues used several tricks to see just what NFkB is up to in the brain and how its activity relates to ageing. By linking it to a fluorescent molecule, the authors were able to visualise NFkB activity in the brain. The more active the transcription factor, the brighter the signal. In this case, brains got brighter with age supporting the idea that inflammation in the brain increases as we get older. While not always bad, inflammation in the brain is a sign of altered equilibrium and contributes to several neurodegenerative diseases3-6. By injecting a non-functional version of the protein, the authors were able to interrupt NFkB activation in the brains of old mice. Interestingly, reducing NFkB activity not only reduced inflammation, but also improved cognitive function (measured by maze navigation), decreased muscle degeneration (measured by grip tests) and even extended life span.
So why is inflammation increased in older brains? It turns out that in mouse middle age, the majority of NFkB activity comes from the microglia –the brain’s resident immune cells. A change in the calibration of these cells over time increases their sensitivity in old age. Instead of waiting for an infection, the microglia are active all the time and mount an inappropriate inflammatory response. This is when NFkB first gets activated and it makes the microglia produce other inflammatory molecules, which perpetuate the rumour that there is some kind of infection and that inflammation is necessary. Neurons take up these signals from the microglia and NFkB inside these cells also becomes active. This form of molecular baton passing between the immune microglia and the neurons of the hypothalamus is possible because they share the same signalling molecules, and is likely to bounce back and forth in an escalating feedback loop. In this way, an inappropriate response by one cell type (the microglia) can result in another cell type getting the wrong idea. As the signals feedback, the neurons change their behaviour. One consequence is that they gradually reduce production of an important molecule called gonadotropin-releasing hormone (GnRH). The decline of GnRH is responsible (along with other things) for the loss of reproductive function with age; but this new study showed that GnRH injection can also improve neural regeneration in old mice and strengthen cognitive and muscle function. In addition, by specifically removing a gene controlling NFkB in microglia, the authors were able to restrain inflammatory microglia in middle-aged mice. This nipped inflammation in the bud, prevented the involvement of neurons in the inflammatory process, reduced GnRH decline and extended lifespan. Hurray! A new miracle drug, right? Well, maybe not quite yet.
Unfortunately, we’re still a long way from using NFkB inhibition or GnRH replacement to extend human lifespan. The fact that many cells are using the same molecules to transmit messages makes targeting specific molecules very difficult. During evolution, different cell types adopted different roles as they became more specialised. However, having come from the same ancestral cell, they use the same old machinery to perform new and different tasks. Now, when a signal from one type of cell gets picked up by another, there is a level of cross-communication that can have unexpected and sometimes undesirable outcomes. If we try and remove a protein that is misbehaving in one cell type, we risk interrupting its routine function in another. The complex crosstalk between biological molecules is a caveat worth remembering whenever a new protein or gene hits the headlines as the root of all ageing or instigator of disease. These genes and proteins did not evolve to make us old and sick. They evolved to perform a task that is almost certainly essential for survival. It is the misuse of a protein or its appearance in the wrong cell at the wrong time that causes problems. A “disease-causing” protein is, in all probability, performing an essential role elsewhere in the body so cannot just be eliminated.
It will take incredibly sophisticated techniques to iron out exactly how best to recalibrate cellular function following an undesirable change like the increase in NFkB activity in older neurons. Unravelling the mysteries of ageing is not an easy task and is likely to involve the integration of many different scientific disciplines. By identifying how different cells and organs communicate, we hope to get a better idea of when and how signalling wires get crossed and so identify the right target to prevent disease and maybe even slow the ageing process.
1) Alzheimer’s Association (2013). 2012 Alzheimer’s Disease Facts and Figures. http://www.alz.org/downloads/facts_figures_2012.pdf OPEN ACCESS!
2) Zhang et al. (2013). Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 497: 211-216.
3) Wright et al. (2013) Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE 8(4): e59586. http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0059586 OPEN ACCESS!
4) Ugur Tufekci et al. (2012). Chapter Four – Inflammation in Parkinson's Disease. Advances in Protein Chemistry and Structural Biology Vol. 88 pp. 69–132. http://dx.doi.org/10.1016/B978-0-12-398314-5.00004-0
5) Holmes (2013). Systemic inflammation and Alzheimer's disease. Neuropathology and Applied Neurobiology 39 (1): 1365-2990 http://dx.doi.org/10.1111/j.1365-2990.2012.01307.x
6) Hauser and Oksenberg (2006). The Neurobiology of Multiple Sclerosis: Genes, Inflammation, and Neurodegeneration. Neuron 52 (1): 61–76 http://dx.doi.org/10.1016/j.neuron.2006.09.011